CE marking is equivalent to a standardisation mark, and it is affixed to manufactured products that comply with European standards.
What is CE Marking Certification?
CE stands for “Conformité Européenne,” and it is your certification that your product meets the essential Requirements of the relevant European Legislation. You will have immediate access to all EU and EEA markets, as well as any other international markets where CE Marking is approved. The following product categories applies to at least one of the CE marking directives:
- Medical devices
- Active implantable medical devices
- In vitro diagnostic medical devices
Previously, CE certification was limited to the electronic items sold within the European Economic Area (EEA). But as time progresses, the CE marking extends its footprint to the rest of the world. It should be noted that the CE mark is not considered as a benchmark for quality for electronic products.
- The product is being manufactured as per European product directives.
- The product meets European safety and quality standards.
- The product is free from environmental and health hazards.
- It is free to move within the European Free Trade Association (EFTA) and the European Union (EU).
- Allows commercialisation across all 30 countries in the EEA
- Provides credibility to your organization in the eyes of your patients and customers
- Demonstrates you have implemented a QMS and can maintain documentation quality
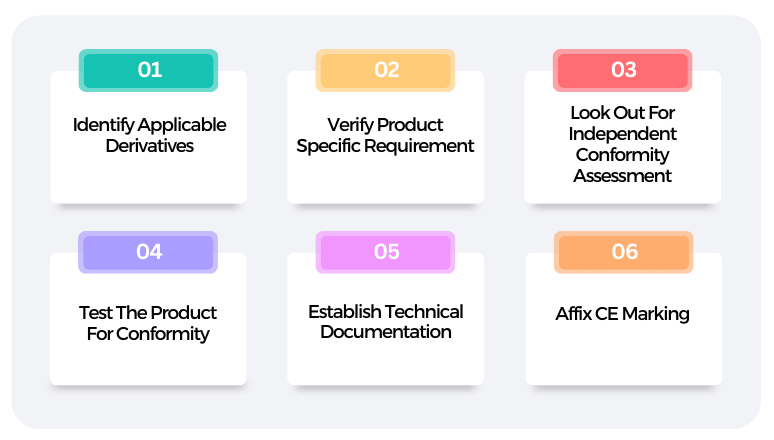
The CE Mark Process: Step-by-step approach
The 10 steps below illustrate the CE approval process in Europe.
Step 1
To obtain CE Marking certification, you must comply with European Commission Regulation (EU) No. 2017/746, commonly known as the In Vitro Device Regulation (IVDR).
Step 2
Appoint a Person Responsible for regulatory compliance. Determine classification of your device using Annex VIII (Classification Criteria) of the IVDR* – Class A (Non-sterile); Class A (sterile); Class B, Class C, or Class D.
Step 3
For all devices except Class A (non-sterile), implement a Quality Management System (QMS) in accordance with the IVDR. Most companies apply the EN ISO 13485 standard to achieve compliance. Your QMS must include Performance Evaluation, Post-Market Surveillance (PMS) and Post Market Performance Follow-up (PMPF) plans. Plan in advance with suppliers about unannounced Notified Body audits.
For Class A (non-sterile), you must implement a QMS though Notified Body intervention is not required.
Step 4
In accordance with Annex II and III of the IVDR, prepare a CE Technical File or Design Dossier (Class III) providing information about your device and its intended use plus testing reports, Performance Evaluation Plan, risk management file, IFU, labelling and more. Obtain a Unique Device Identifier (UDI) for your device.
Step 5
If you do not have a location in Europe, appoint an Authorized Representative (EC REP) located in the EU who is qualified to handle regulatory issues. Place your EC REP name and address on device label. Obtain a Single Registration Number from the regulators.
Step 6
For all devices except Class A (non-sterile), your QMS and Technical File or Design Dossier must be audited by a Notified Body, a third party accredited by European authorities to audit medical device companies and products.
Step 7
For all devices except Class A (non-sterile), you will be issued a European CE Marking Certificate for your device and an ISO 13485 certificate for your facility following successful completion of your Notified Body audit. ISO 13485 certification must be renewed every year. CE Marking certificates are typically valid for a maximum of 5 years but are typically reviewed during your annual surveillance audit.
Step 8
Prepare a Declaration of Conformity in accordance with Annex IV, a legally binding document prepared by the manufacturer stating that the device is in compliance with the applicable European requirements. You may now affix the CE Marking.
Step 9
Register the device and its Unique Device Identifier (UDI) in the EUDAMED database. UDI must be on label and associated with the regulatory documents.
Step 10
For Class A (non-sterile), annual NB audits are not required. However, your Performance Evaluation Report, Technical File, and PMS activities must be kept updated.
For all other classes, you will be audited each year by a Notified Body to ensure ongoing compliance with the IVDR. Failure to pass the audit will invalidate your CE Marking certificate. You must perform Performance Evaluation, PMS, and PMPF activities to maintain certification.
Did you know?
We are able to support you with achieving CE Mark certification as part our full-service medical kitting solution.